--min-end-score
This threshold is applied to the two individual barcode scores, the leading and trailing barcode. ZMWs with at least one individual barcode score below N are omitted. The default is 0.
Simplified example: A ZMW is tagged with two barcodes A and B. All leading barcode regions match to A with score 90 and all trailing barcode regions match B with score 30. On average, the barcode score is (90+30)/2=60. Option --min-end-score filters on an individual barcode level, checking 90 and 30. Using --min-end-score 45, this ZMW would not pass, because B is below the threshold.
This filter can be used to remove ZMWs that have one good and one bad call, only useful for asymmetric barcoding schemes with different barcodes in a pair. For libraries with the same barcode in pair, this option is identical to --min-score.
--min-ref-span && --min-scoring-regions
Those options are used in combination to remove ZMWs that have spurious barcode hits. Option --min-ref-span defines the minimum reference (read) span relative to the barcode length to call a barcode region scoring.
Option --min-scoring-regions defines the minimum number of scoring barcode regions. ZMWs with less than -min-scoring-regions N scoring regions are omitted.
The following schematic shows a simplified example, in which the relative reference spans are 0.4 for leading and 0.8 for trailing barcode regions, as the barcodes are 10 bp long and span with 4 bp and 8 bp. If we set --min-ref-span 0.5 then only the leading barcode region would be flagged as scoring. Requiring --min-scoring-regions 2, this ZMW would fail, as only one region is above the required reference span. 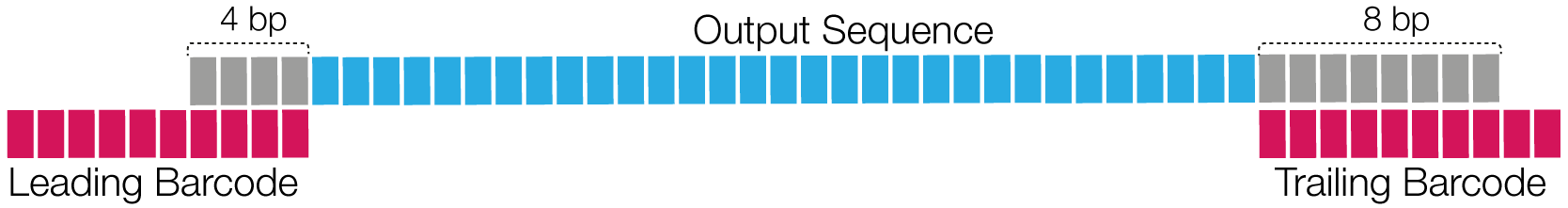
Why do most of my ZMWs get filtered by the score lead threshold?
The score lead measures how close the best barcode call is to the second best. Possible solutions without seeing your data:
- Is that sample actually barcoded?
- Are your barcode sequences genetically too close for SMRT sequencing? Try CCS calling first and demultiplex with
--ccs. - Are the synthesized products clean and not degenerate?
- Did the sequencing run perform optimally, is the accuracy in the expected range?
- Did you run lima twice, first on the original and then on the already demultiplexed data? This is not supported, as the barcodes have been clipped and removed.
Try to decrease --score-lead, with the potential risk of introducing false positives.
--max-scored-barcode-pairs
Only use up to first N barcode pair regions for barcode identification. The default is 0, deactivated filter. This is equivalent to the maximum number of scored adapters in bam2bam.
--max-scored-barcodes
Only use up to first N barcode regions for barcode identification. The default is 0, deactivated filter. Setting to 1 enables single-pass and single-barcode calculation.
--max-scored-adapters
Only use the flanking regions of up to first N adapters for barcode identification. Only full adapters, surrounded by subreads, are used. The default is 0, deactivated filter. Setting to 1 enables single-pass barcode calculation using forward and reverse pass.
Caution: If a subread between two full flanking adapters gets removed in PPA, lima will score frankstein flanking barcodes for two consecutive subreads with what it believes one adapter in the middle.
--score-full-pass
Only use reads flanked by adapters on both sides for barcode identification, full-pass reads. This is implicit for CCS reads.
--per-read
Score and tag per subread, instead per ZMW. This is implicit for CCS reads.
--single-side
Identify barcodes in molecules that only have barcodes adjacent to one adapter. This approach makes no assumption about an alternating pattern of barcoded and barcode-free adapters. In contrast, a 1D k-means similar to the original Lloyd algorithm is employed to identify two clusters to separate low- and high-scoring barcode regions. This method does not suffer from irregular adapter calls, but the additional flexibility might lead to yet-unknown problems.
For this mode, high-scoring barcode regions are whitelisted. Only whitelisted barcode regions contribute to the final mean barcode score and to the calculation of --min-score-lead.
--scored-adapter-ratio
Minimum ratio of scored vs sequenced adapters. The default is 0.25.
--enforce-first-barcode
Set the first barcode to be barcode index 0.